本文介绍如何使用E-HPC集群运行BWA、GATK、Samtools软件进行基因测序计算。
背景信息
生命科学领域内基因测序技术的飞速发展,人类发现的基因序列以指数级增长,对于如此数量庞大的基因进行同源性搜寻、比对、变异检查等,往往伴随着巨大的数据处理和并行计算。高性能计算可以提供强大的算力支持,使用多种调度器提高并发效率,使用GPU进行计算加速等。
本文以经典及普及的二代全基因组测序WGS(Whole Genome Sequencing)流程为例,结合二代测序软件GATK,介绍人类全基因组测序的通用流程。在实际生信分析中,需要结合不同的业务需求进行相应的调整,如增加变异检测质控及过滤等,您可以结合实际场景进行调整。
在进行基因测序时,您可以使用BWA构建索引及比对记录,再使用Samtools对比对记录进行排序,然后使用GATK去除重复序列、重新校正碱基质量值、变异检查。
BWA(Burrows-Wheeler-Alignment Tool)是一款将DNA序列映射到参考基因组上的软件,例如比对人类基因组。
GATK(The Genome Analysis Toolkit)是一款二代重测序数据分析软件,是基因分析的工具集。主要用于去除重复序列、重新校正碱基质量值、变异检查等。
Samtools是用于处理sam和bam格式的工具软件,能够查看二进制文件、转换文件格式、对文件排序及合并,可以结合sam格式文件中的flag、tag等信息,对比对结果进行统计汇总。
准备工作
创建E-HPC集群。具体操作,请参见使用向导创建集群。
配置集群时,软硬件参数配置如下:
参数
说明
硬件参数
部署方式为精简,包含1个管控节点和1个计算节点,规格如下:
管控节点:采用ecs.c7.large实例规格,该规格配置为2 vCPU,4 GiB内存。
计算节点:采用ecs.ebmc5s.24xlarge实例规格,该规格配置为96 vCPU、192 GiB内存。
软件参数
镜像选择CentOS 7.6公共镜像,调度器选择pbs,打开VNC开关。
创建集群用户。具体操作,请参见创建用户。
集群用户用于登录集群,进行编译软件、提交作业等操作。本文创建的用户示例如下:
用户名:testuser
用户组:sudo权限组
安装软件。
步骤一:连接集群
选择以下一种方式连接集群:
通过客户端
该方式仅支持使用PBS调度器的集群。操作前,请确保您已下载安装E-HPC客户端,且已配置客户端所需环境。具体操作,请参见配置客户端所需环境。
打开并登录E-HPC客户端。
在客户端左侧导航栏,单击会话管理。
在会话管理页面的右上角,单击terminal,打开Terminal窗口。
通过控制台
登录弹性高性能计算控制台。
在顶部菜单栏左上角处,选择地域。
在左侧导航栏,单击集群。
在集群页面,找到目标集群,单击远程连接。
在远程连接页面,输入集群用户名、登录密码和端口,单击ssh连接。
步骤二:进行基因测序
切换到计算节点。
以compute000节点为例:
ssh compute000(可选)下载tmux并创建一个tmux session。
说明基因测序的流程包括构建索引、比对、排序、去重、创建字典、构建校准表、重校准和变异检查等,整体需要6~7小时,建议您在tmux session中执行,以免因ECS断开连接而中断操作。各阶段的具体耗时可参考测序耗时说明。
sudo yum install tmux tmux准备基因测序需要的基因文件。
下载并解压人类参考基因组、人类基因测序样本1、人类基因测序样本2。
wget https://public-ehpc-package.oss-cn-hangzhou.aliyuncs.com/lifescience/b37_human_g1k_v37.fasta wget https://public-ehpc-package.oss-cn-hangzhou.aliyuncs.com/lifescience/b37_1000G_phase1.indels.b37.vcf wget https://public-ehpc-package.oss-cn-hangzhou.aliyuncs.com/lifescience/b37_Mills_and_1000G_gold_standard.indels.b37.vcf wget https://public-ehpc-package.oss-cn-hangzhou.aliyuncs.com/lifescience/b37_dbsnp_138.b37.vcf.zip unzip b37_dbsnp_138.b37.vcf.zip wget https://public-ehpc-package.oss-cn-hangzhou.aliyuncs.com/lifescience/gatk-examples_example1_NA20318_ERR250971_1.filt.fastq.gz gunzip gatk-examples_example1_NA20318_ERR250971_1.filt.fastq.gz wget https://public-ehpc-package.oss-cn-hangzhou.aliyuncs.com/lifescience/gatk-examples_example1_NA20318_ERR250971_2.filt.fastq.gz gunzip gatk-examples_example1_NA20318_ERR250971_2.filt.fastq.gz对下载解压的基因文件进行压缩并建立索引。
使用bgzip对文件进行压缩。
bgzip -f b37_1000G_phase1.indels.b37.vcf bgzip -f b37_Mills_and_1000G_gold_standard.indels.b37.vcf bgzip -f b37_dbsnp_138.b37.vcf使用tabix建立索引。
tabix -p vcf b37_1000G_phase1.indels.b37.vcf.gz tabix -p vcf b37_Mills_and_1000G_gold_standard.indels.b37.vcf.gz tabix -p vcf b37_dbsnp_138.b37.vcf.gz
构建索引。
构建BWA比对所需的参考基因组的index数据库。
time bwa index b37_human_g1k_v37.fasta预期返回如下:
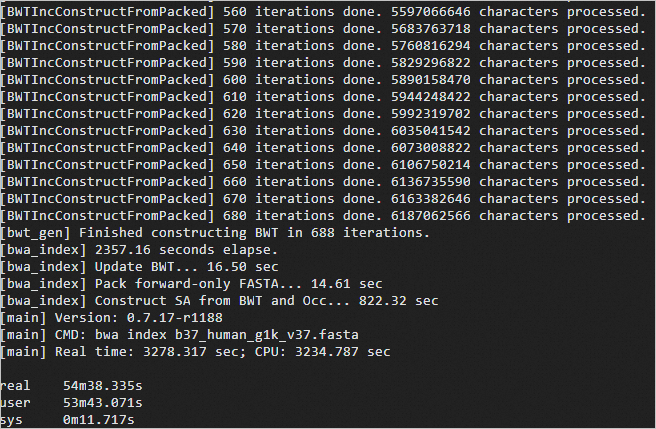
创建fasta序列格式索引。
samtools faidx b37_human_g1k_v37.fasta
比对reads。
将样本测序数据reads与人类参考基因组进行比对,并将输出文件转化为bam格式,以节省磁盘空间。
time bwa mem -t 52 \ -R '@RG\tID:ehpc\tPL:illumina\tLB:library\tSM:b37' \ b37_human_g1k_v37.fasta \ gatk-examples_example1_NA20318_ERR250971_1.filt.fastq \ gatk-examples_example1_NA20318_ERR250971_2.filt.fastq | samtools view -S -b - > ERR250971.bam预期返回如下:
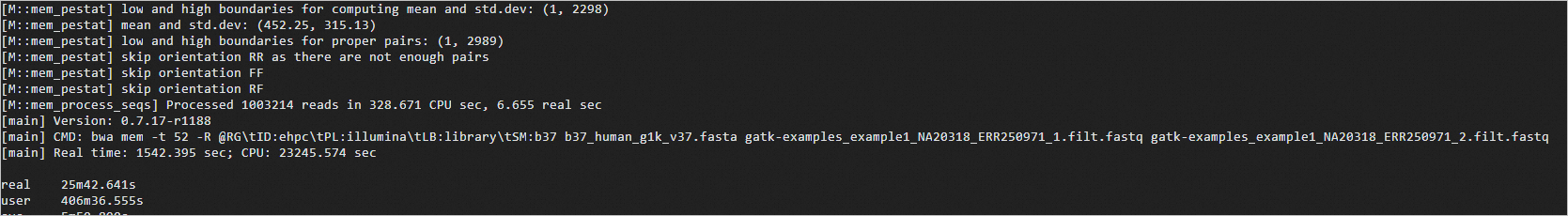
将比对记录按照顺序从小到大进行排序。
time samtools sort -@ 52 -O bam -o ERR250971.sorted.bam ERR250971.bam去除重复序列。
去除DNA序列PCR扩增产生的重复reads序列。
time gatk MarkDuplicates \ -I ERR250971.sorted.bam -O ERR250971.markdup.bam \ -M ERR250971.sorted.markdup_metrics.txt预期返回如下:
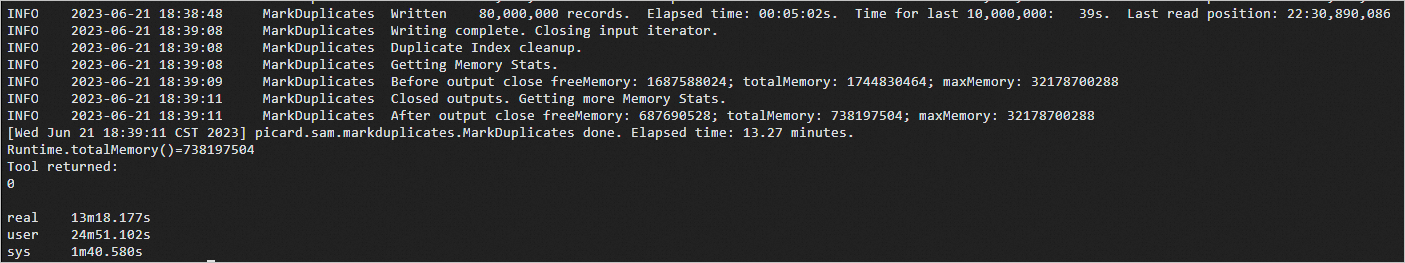
构建markdup测序bam文件索引。
samtools index ERR250971.markdup.bam
生成参考基因组dict文件。
time gatk CreateSequenceDictionary \ -R b37_human_g1k_v37.fasta \ -O b37_human_g1k_v37.dict重新校正碱基质量值。
利用已有的snp及indels数据库,建立相关模型,构建重校准表。
time gatk BaseRecalibrator \ -R b37_human_g1k_v37.fasta \ -I ERR250971.markdup.bam \ --known-sites b37_1000G_phase1.indels.b37.vcf.gz \ --known-sites b37_Mills_and_1000G_gold_standard.indels.b37.vcf.gz \ --known-sites b37_dbsnp_138.b37.vcf.gz -O ERR250971.BQSR.table预期返回如下:
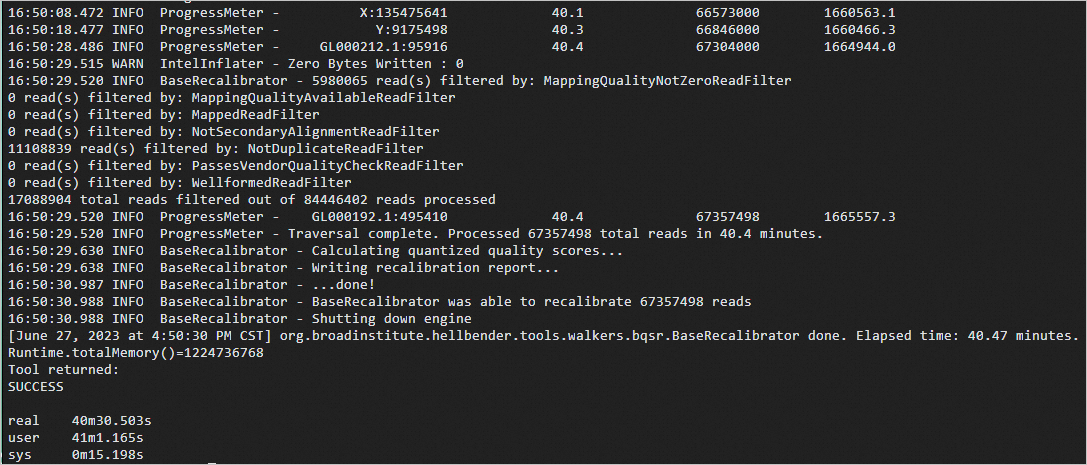
对原始碱基质量值进行调整。
time gatk ApplyBQSR \ --bqsr-recal-file ERR250971.BQSR.table \ -R b37_human_g1k_v37.fasta \ -I ERR250971.markdup.bam \ -O ERR250971.BQSR.bam预期返回如下:
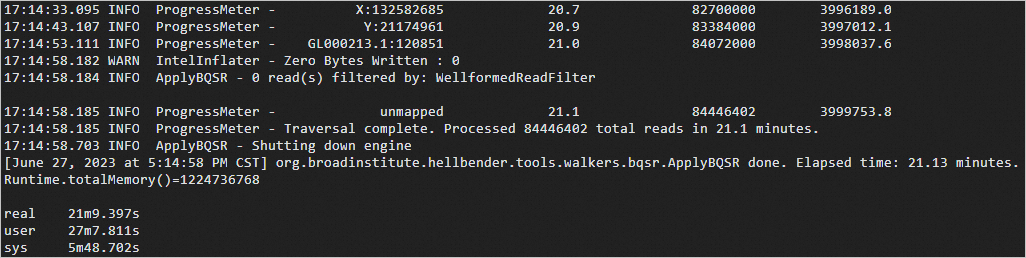
构建BQSR测序bam文件索引。
time samtools index ERR250971.BQSR.bam
输出变异检测结果vcf文件。
time gatk HaplotypeCaller \ -R b37_human_g1k_v37.fasta \ -I ERR250971.BQSR.bam \ -O ERR250971.HC.vcf.gz预期返回如下:
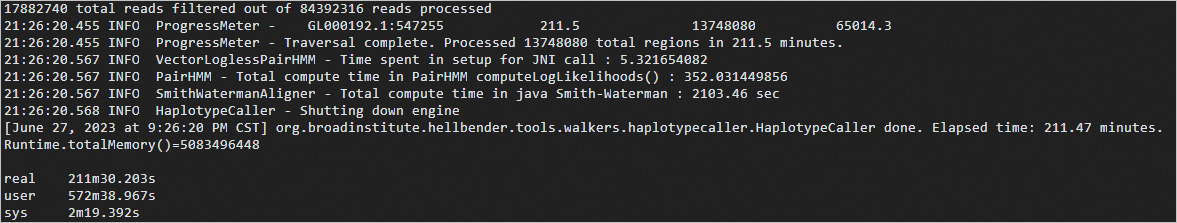
测序耗时说明
本文使用的计算节点为ecs.ebmc5s.24xlarge,仅为演示全基因组测序流程,整体大约耗时6~7小时,下表展示了基因测序各流程大约使用的时间供您参考,并非最优性能。
流程 | 功能 | 运行时间(min) |
bwa index | 构建索引 | 54 |
bwa mem | 比对 | 25 |
samtools sort | 排序 | 2 |
gatk MarkDuplicates | 去重 | 13 |
gatk CreateSequenceDictionary | 创建字典 | 0.15 |
gatk BaseRecalibrator | 构建校准表 | 40 |
gatk ApplyBQSR | 重校准 | 21 |
gatk HaplotypeCaller | 变异检查 | 211 |
- 本页导读 (1)