In the highly complex and data-intensive field of genetic computation, researchers and bioinformaticians face significant challenges, including managing explosive data growth and efficiently integrating and analyzing data to uncover biological insights. To meet these challenges, workflow automation and orchestration have become key technologies. Among them, Argo Workflows stands out for its containerization, flexibility, and ease of use, making it a powerful tool for connecting the various stages of genetic computation. This topic describes how to orchestrate a genetic computation workflow by using Argo Workflows.
Background
Genetic computation workflows
A genetic computation workflow is a process that organizes interconnected computing tasks and data processing steps in a defined order to achieve a specific analysis goal in genomics research. These workflows typically include complex steps such as data preprocessing, sequence alignment, variant detection, gene expression analysis, and phylogenetic tree construction.
Argo Workflows for genetic computation workflows
Argo Workflows is an open-source, Kubernetes-native workflow engine designed for containerized environments that orchestrates complex workflows with flexibility and efficiency. The advantages of Argo Workflows are particularly significant in genetic computation scenarios.
-
Containerization and environmental consistency: Genetic analysis involves numerous software tools and dependency libraries. By encapsulating each analysis step into a Docker container, Argo Workflows ensures cross-platform consistency and reproducibility, reducing reproducibility issues across different environments.
-
Flexible orchestration capabilities: Workflows in genomics research often involve multiple steps, conditional branches, and parallel processing requirements. Argo Workflows supports complex logic and conditional controls, making it simple and intuitive to customize workflows as needed.
Although open-source Argo Workflows offers significant advantages in orchestrating genetic computation workflows, users face several practical challenges.
-
Large-scale operational challenges: Faced with a massive number of tasks, users with a research background may lack deep experience in cluster operations and maintenance. This makes it difficult to implement effective cluster optimization and maintenance strategies.
-
Complex workflow orchestration challenges: Scientific experiments often involve vast parameter spaces and numerous process steps, often involving thousands of jobs. Open-source workflow engines struggle to support this scale.
-
Resource optimization and elastic scaling difficulties: Genetic data analysis is typically computationally intensive. Users want to intelligently schedule resources based on workload to achieve high resource utilization and support automatic scaling of computing power on demand. Open-source solutions often fall short in meeting these needs.
To address challenges in genetic computation, such as large-scale operations, complex workflow orchestration, and resource optimization with elastic scaling, the Alibaba Cloud ACK One team has introduced the distributed workflow Argo cluster.
Distributed workflow Argo cluster
The Alibaba Cloud distributed workflow Argo cluster is based on the open-source Argo Workflows project. It adopts a serverless model and runs workflows on Elastic Container Instance (ECI). By optimizing Kubernetes cluster parameters, the solution achieves efficient elastic scheduling for large-scale workflows and optimizes costs by using spot instances of ECI. This solution supports various execution strategies such as concurrency, loops, and retries, making it suitable for typical genetic computation processes and highly complex workflow orchestration tasks.

With its containerized design, flexible orchestration, and simple operation, Argo Workflows excels in genetic computation and other data-intensive research fields, significantly improving automation, resource utilization, and analysis efficiency. The Alibaba Cloud ACK One team is one of the pioneers in applying Argo Workflows for large-scale task orchestration and has accumulated extensive best practices in scenarios such as genetic computation, autonomous driving, and financial simulation. Contact the ACK One team by joining the DingTalk group (ID: 35688562) to exchange ideas and insights.
Orchestrate a genetic computation workflow
This example uses a classic Burrows-Wheeler Aligner (BWA) sequence alignment pipeline to demonstrate how to edit and run a genetic computation workflow using Argo Workflows.
-
Mount an Alibaba Cloud OSS storage volume, allowing the workflow to operate on files in OSS as if they were local files. For more information, see Use storage volumes.
-
Use the following YAML to create a workflow. For more information, see Create a workflow.
The process consists of three main stages:
-
bwaprepare: The data preparation stage. Downloads and decompresses the fastq and reference files, and builds an index for the reference genome. -
bwamap: The sequence alignment stage. Aligns the sequencing data with the reference genome and generates alignment results. This stage processes multiple files in parallel. -
bwaindex: The indexing stage. Generates, sorts, and indexes the BAM file produced by aligning raw sequencing data with the reference genome, and provides a view of the alignment results.
-
-
After the workflow runs, you can view the task's Directed Acyclic Graph (DAG) and the run results in the Workflow Console (Argo).
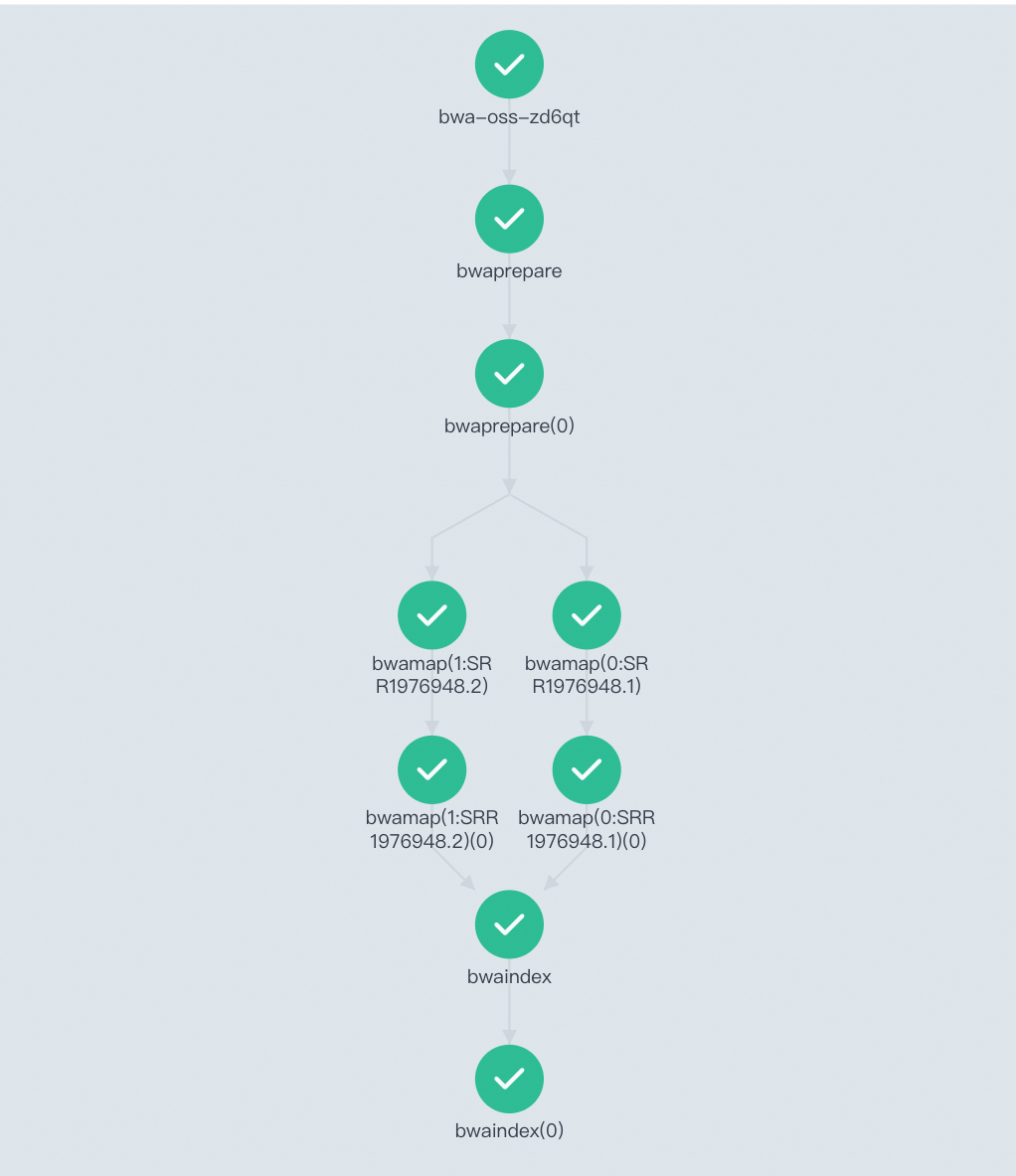
You can also log on to the OSS console to find the generated alignment result files in the corresponding OSS folder.
The process generates 17 files in total. The generated files include the input FASTQ files (
SRR1976948_1.fastq.gzandSRR1976948_2.fastq.gz), intermediate BWA alignment files (.saiand.sam), the final BAM result files (.bam,.sorted.bam, and.bai), and the reference genomesubset_assembly.fawith its index files (.amb,.ann,.bwt,.fai,.pac, and.sa).
Related documents
-
For more information about the distributed workflow Argo cluster, see Overview of the distributed workflow Argo cluster.
-
To learn more about Argo Workflows, see the open-source Argo Workflows documentation.