在基因计算这一高度复杂且数据密集型的领域,科研人员和生物信息分析师面临着严峻挑战,这不仅体现在数据量的爆炸性增长上,还在于需要高效、准确地整合和分析数据,以揭示生命的奥秘。为应对这些挑战,工作流自动化编排成为关键技术,其中,Argo Workflows以其容器化、灵活性和易用性脱颖而出,成为串联基因计算各个环节的得力助手。本文介绍如何使用Argo Workflows编排基因计算工作流。
背景信息
基因计算工作流
基因计算工作流是在基因组学研究中,为实现特定分析目标,将多个互相关联的计算任务和数据处理步骤有序组织成的执行流程。这些工作流通常涵盖数据预处理、序列比对、变异检测、基因表达分析和进化树构建等复杂步骤。
Argo Workflows基因计算工作流
Argo Workflows是一个开源的Kubernetes原生工作流引擎,专为容器化环境设计,能够灵活、高效地编排复杂的工作流程。在基因计算场景中,Argo的优势尤为显著。
尽管开源Argo Workflows在基因计算工作流的编排领域展现了显著的优势,但在实际应用中仍需克服若干挑战。
-
大规模运维难题:面对庞大的任务规模,加之科研背景的用户可能缺乏深度的集群运维经验,难以实施高效的集群优化与维护策略。
-
复杂工作流编排挑战:科研实验的特性决定了参数空间的广阔及流程步骤的繁多,往往涉及成千上万的作业项,开源工作流引擎难以支持。
-
资源优化与弹性伸缩困难:基因数据分析通常消耗大量计算资源,用户希望能够根据工作负载智能调度资源,实现资源的高效利用,并支持按需自动扩展计算能力,开源方案难以满足。
为应对基因计算中大规模运维、复杂工作流编排、资源优化与弹性伸缩等挑战,阿里云ACK One团队推出了分布式工作流Argo集群。
分布式工作流Argo集群
阿里云分布式工作流Argo集群基于开源项目Argo Workflows实现,采用无服务器模式,使用阿里云弹性容器实例ECI运行工作流,通过优化Kubernetes集群参数,实现大规模工作流的高效弹性调度,并结合抢占式ECI实例,优化成本。该解决方案支持并发、循环、重试等多种执行策略,适用于典型的基因计算过程和高度复杂的工作流任务编排。

说明
Argo Workflows凭借其容器化设计、灵活编排和简易操作,在基因计算及其他数据密集型科研领域表现出色,显著提升了自动化水平、资源利用率和分析效率。阿里云ACK One团队是较早在大规模任务编排中应用Argo Workflows的团队之一,并在基因计算、自动驾驶、金融模拟等场景积累了丰富的最佳实践经验。欢迎联系ACK One团队,加入钉钉群号35688562,共同交流探讨。
使用Argo Workflows编排基因计算工作流
本示例通过一个经典的BWA测序比对流程,展示如何使用Argo Workflows来编辑并执行基因计算工作流。
-
创建分布式工作流Argo集群。
-
开启Argo Server访问工作流集群。
-
挂载阿里云OSS存储卷,使工作流能够像操作本地文件一样操作OSS上的文件。具体操作,请参见使用存储卷。
-
使用以下YAML创建一个工作流。具体操作,请参见创建工作流。
流程主要包括三个阶段:
-
bwaprepare: 数据准备阶段,下载并解压fastq、reference文件,对参考基因组建立索引。
-
bwamap:将测序数据与参考基因组进行比对并生成对齐结果,该阶段并行处理多个文件。
-
bwaindex:从原始测序数据比对到参考基因组,生成、排序、索引BAM文件,并浏览比对结果。
展开查看YAML示例
apiVersion: argoproj.io/v1alpha1
kind: Workflow
metadata:
generateName: bwa-oss-
spec:
entrypoint: bwa-oss
arguments:
parameters:
- name: fastqFolder # 下载文件的保存路径。
value: /gene
- name: reference # 参考基因组文件。
value: https://ags-public.oss-cn-beijing.aliyuncs.com/alignment/subset_assembly.fa.gz
- name: fastq1 # 原始测序数据。
value: https://ags-public.oss-cn-beijing.aliyuncs.com/alignment/SRR1976948_1.fastq.gz
- name: fastq2
value: https://ags-public.oss-cn-beijing.aliyuncs.com/alignment/SRR1976948_2.fastq.gz
volumes: # 远端存储路径挂载。
- name: ossdir
persistentVolumeClaim:
claimName: pvc-oss
templates:
- name: bwaprepare # 数据准备阶段,下载并解压fastq、reference文件,对参考基因组建立索引。
container:
image: registry.cn-beijing.aliyuncs.com/geno/alltools:v0.2
imagePullPolicy: Always
command: [sh,-c]
args:
- mkdir -p /bwa{{workflow.parameters.fastqFolder}}; cd /bwa{{workflow.parameters.fastqFolder}}; rm -rf SRR1976948*;
wget {{workflow.parameters.reference}};
wget {{workflow.parameters.fastq1}};
wget {{workflow.parameters.fastq2}};
gzip -d subset_assembly.fa.gz;
gunzip -c SRR1976948_1.fastq.gz | head -800000 > SRR1976948.1;
gunzip -c SRR1976948_2.fastq.gz | head -800000 > SRR1976948.2;
bwa index subset_assembly.fa;
volumeMounts:
- name: ossdir
mountPath: /bwa
retryStrategy: # 重试机制。
limit: 3
- name: bwamap # 将测序数据与参考基因组进行比对预处理并生成对齐结果。
inputs:
parameters:
- name: object
container:
image: registry.cn-beijing.aliyuncs.com/geno/alltools:v0.2
imagePullPolicy: Always
command:
- sh
- -c
args:
- cd /bwa{{workflow.parameters.fastqFolder}};
bwa aln subset_assembly.fa {{inputs.parameters.object}} > {{inputs.parameters.object}}.untrimmed.sai;
volumeMounts:
- name: ossdir
mountPath: /bwa
retryStrategy:
limit: 3
- name: bwaindex # 从原始测序数据比对到参考基因组,生成、排序、索引BAM文件,并浏览比对结果。
container:
args:
- cd /bwa{{workflow.parameters.fastqFolder}};
bwa sampe subset_assembly.fa SRR1976948.1.untrimmed.sai SRR1976948.2.untrimmed.sai SRR1976948.1 SRR1976948.2 > SRR1976948.untrimmed.sam;
samtools import subset_assembly.fa SRR1976948.untrimmed.sam SRR1976948.untrimmed.sam.bam;
samtools sort SRR1976948.untrimmed.sam.bam -o SRR1976948.untrimmed.sam.bam.sorted.bam;
samtools index SRR1976948.untrimmed.sam.bam.sorted.bam;
samtools tview SRR1976948.untrimmed.sam.bam.sorted.bam subset_assembly.fa -p k99_13588:1000 -d T;
command:
- sh
- -c
image: registry.cn-beijing.aliyuncs.com/geno/alltools:v0.2
imagePullPolicy: Always
volumeMounts:
- mountPath: /bwa/
name: ossdir
retryStrategy:
limit: 3
- name: bwa-oss # 各阶段流程编排。
dag:
tasks:
- name: bwaprepare # 先执行数据准备。
template: bwaprepare
- name: bwamap # 预处理,并生成对齐结果。
template: bwamap
dependencies: [bwaprepare] # 依赖数据准备阶段。
arguments:
parameters:
- name: object
value: "{{item}}"
withItems: ["SRR1976948.1","SRR1976948.2"] # 并行处理多个文件。
- name: bwaindex # 比对并浏览比对结果。
template: bwaindex
dependencies: [bwamap] # 依赖预处理阶段。
-
工作流运行后,您可以工作流控制台(Argo)查看任务DAG流程与运行结果。
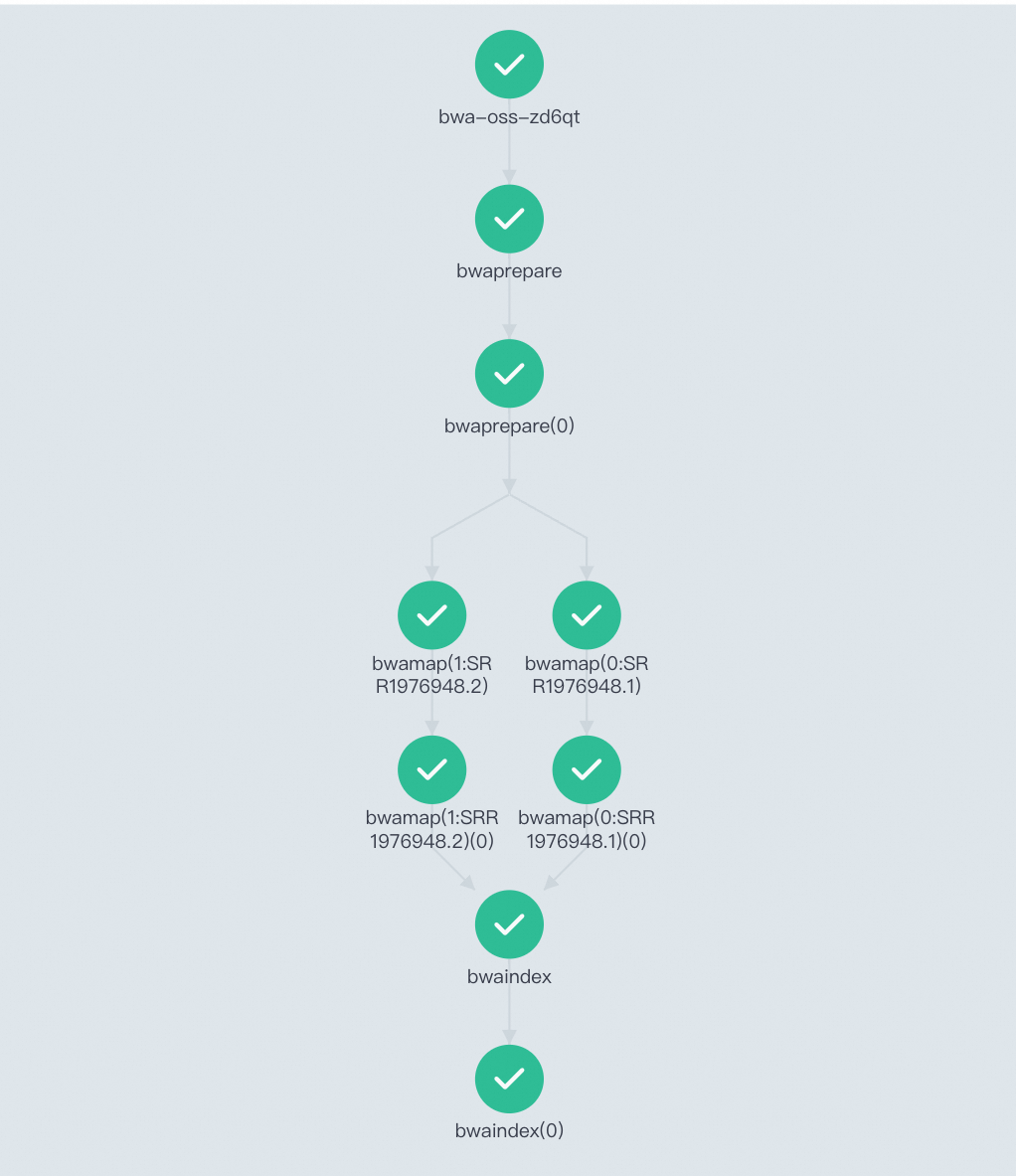
也可以通过登录OSS管理控制台,在OSS对应文件夹中发现比对结果文件已成功生成。
生成的文件包括输入FASTQ文件(SRR1976948_1.fastq.gz、SRR1976948_2.fastq.gz)、BWA比对中间文件(.sai、.sam)、最终BAM结果文件(.bam、.sorted.bam、.bai)以及参考基因组subset_assembly.fa及其索引文件(.amb、.ann、.bwt、.fai、.pac、.sa),共17个文件。
