本文以GROMACS软件为例介绍如何在E-HPC上进行分子动力学模拟。
背景信息
GROMACS(GROningen MAchine for Chemical Simulations)是一款通用软件,用于对具有数百万颗粒子的系统进行基于牛顿运动方程的分子动力学模拟。
GROMACS主要用于生物化学分子,如蛋白质、脂质等具有多种复杂键合相互作用的核酸分析。GROMACS计算典型的模拟应用,如高效地计算非键合相互作用,许多研究人员用其研究非生物系统的聚合物。
GROMACS支持分子动力学的常见算法,可以采用GPU来加速核心计算过程。更多信息,请参见GROMACS官网。
相关算例
算例1:水中的溶菌酶
本算例为一个蛋白质加上离子在水盒子里的模拟过程。
算例下载地址:https://public-ehpc-package.oss-cn-hangzhou.aliyuncs.com/Lysozyme.tar.gz
算例2:水分子运动
本算例为模拟大量水分子在给定空间、温度内的运动过程。
算例下载地址:https://public-ehpc-package.oss-cn-hangzhou.aliyuncs.com/water_GMX50_bare.tar.gz
准备工作
创建E-HPC集群。具体操作,请参见使用向导创建集群。
配置集群时,本文使用的软硬件参数配置示例如下:
参数
说明
硬件参数
部署方式为标准,包含2个管控节点,1个计算节点和1个登录节点,其中计算节点使用GPU实例规格(例如ecs.gn5-c8g1.2xlarge)。
软件参数
镜像选择CentOS 7.2公共镜像,调度器选择pbs,并打开VNC开关。
创建集群用户。具体操作,请参见创建用户。
集群用户用于登录集群,进行编译软件、提交作业等操作,本文创建的用户示例如下:
用户名:gmx.test
用户组:sudo权限组
安装软件。具体操作,请参见安装软件。
需安装的软件如下:
gromacs-gpu,版本为2018.1。
openmpi,版本为3.0.0。
cuda-toolkit,版本为9.0。
vmd,版本为1.9.3。
步骤一:连接集群
选择以下一种方式连接集群。本文使用的用户名为gmx.test,连接集群后会默认登录到/home/gmx.test。
通过客户端
该方式仅支持使用PBS调度器的集群。操作前,请确保您已下载安装E-HPC客户端,且已配置客户端所需环境。具体操作,请参见配置客户端所需环境。
打开并登录E-HPC客户端。
在客户端左侧导航栏,单击会话管理。
在会话管理页面的右上角,单击terminal,打开Terminal窗口。
通过控制台
登录弹性高性能计算控制台。
在顶部菜单栏左上角处,选择地域。
在左侧导航栏,单击集群。
在集群页面,找到目标集群,单击远程连接。
在远程连接页面,输入集群用户名、登录密码和端口,单击ssh连接。
步骤二:提交作业
执行以下命令下载并解压算例。
本文使用水分子运动算例作为示例。
wget https://public-ehpc-package.oss-cn-hangzhou.aliyuncs.com/water_GMX50_bare.tar.gz tar xzvf water_GMX50_bare.tar.gz chown -R gmx.test water-cut1.0_GMX50_bare chgrp -R users water-cut1.0_GMX50_bare执行以下命令创建作业脚本文件,脚本文件命名为gmx.pbs。
vim gmx.pbs作业脚本内容示例如下:
说明本示例使用名为gmx.test的用户提交作业,在一个包含8个vCPU和1块P100 GPU卡的计算节点上运行。实际使用场景中,您可根据集群配置情况做出适当修改。
#!/bin/sh #PBS -j oe #PBS -l select=1:ncpus=8:mpiprocs=4 #PBS -q workq #module命令依赖的环境变量 export MODULEPATH=/opt/ehpcmodulefiles/ module load gromacs-gpu/2018.1 module load openmpi/3.0.0 module load cuda-toolkit/9.0 export OMP_NUM_THREADS=1 cd /home/gmx.test/water-cut1.0_GMX50_bare/0096 #前处理过程,生成tpr格式输入文件 /opt/gromacs-gpu/2018.1/bin/gmx_mpi grompp -f pme.mdp -c conf.gro -p topol.top -o topol_pme.tpr #-ntomp指定每个进程开启的OpenMP线程数,-nsteps指定模拟迭代步数 mpirun -np 4 /opt/gromacs-gpu/2018.1/bin/gmx_mpi mdrun -ntomp 1 -nsteps 100000 -pin on -s topol_pme.tpr执行以下命令提交作业。
qsub gmx.pbs预期返回如下,表示生成的作业ID为0.scheduler。
0.scheduler
步骤三:查看作业结果
查看作业运行情况。
qstat -x 0.scheduler预期返回如下,当返回信息中
S为R时,表示作业正在运行中;当返回信息中S为F时,表示作业已经运行结束。Job id Name User Time Use S Queue ---------------- ---------------- ---------------- -------- - ----- 0.scheduler gmx.pbs gmx.test 00:34:42 F workq说明作业运行需要一定的时间,请您耐心等待。作业运行结束后,您可以执行
cat gmx.pbs.o0查看作业输出。使用VNC可视化查看作业结果。
打开VNC。
说明请确保集群所属安全组已打开VNC所需端口。控制台操作时系统会自动打开12016端口,客户端操作时,请您自行打开端口,首个VNC窗口使用12017端口,如果有多个用户使用VNC,则端口号按顺序递增。
通过客户端
在客户端的左侧导航栏,单击会话管理。
在会话管理页面的右上角,单击VNC,打开VNC窗口。
通过控制台
在弹性高性能计算控制台的左侧导航栏,单击集群。
在集群页面,找到目标集群,单击更多 > VNC。
使用VNC远程连接可视化服务。具体操作,请参见连接可视化服务。
在VNC窗口中,选择Application>System Tools>Terminal。
运行
/opt/vmd/1.9.3/vmd打开VMD软件。在VMD Main对话框中,选择File > New Molecule...。
单击Filename处对应的Browse...,选择结果文件conf.gro。
说明conf.gro文件所在路径为/home/gmx.test/water-cut1.0_GMX50_bare/0096/conf.gro。
单击Load,可在VMD 1.9.3 OpenGL Display窗口查看可视化结果。

步骤四:查看作业计算性能
仅控制台支持查看作业计算性能,客户端暂不支持该功能。
登录弹性高性能计算控制台。
在左侧导航栏,选择作业与性能管理 > E-HPC优化器。
找到目标集群,单击节点。
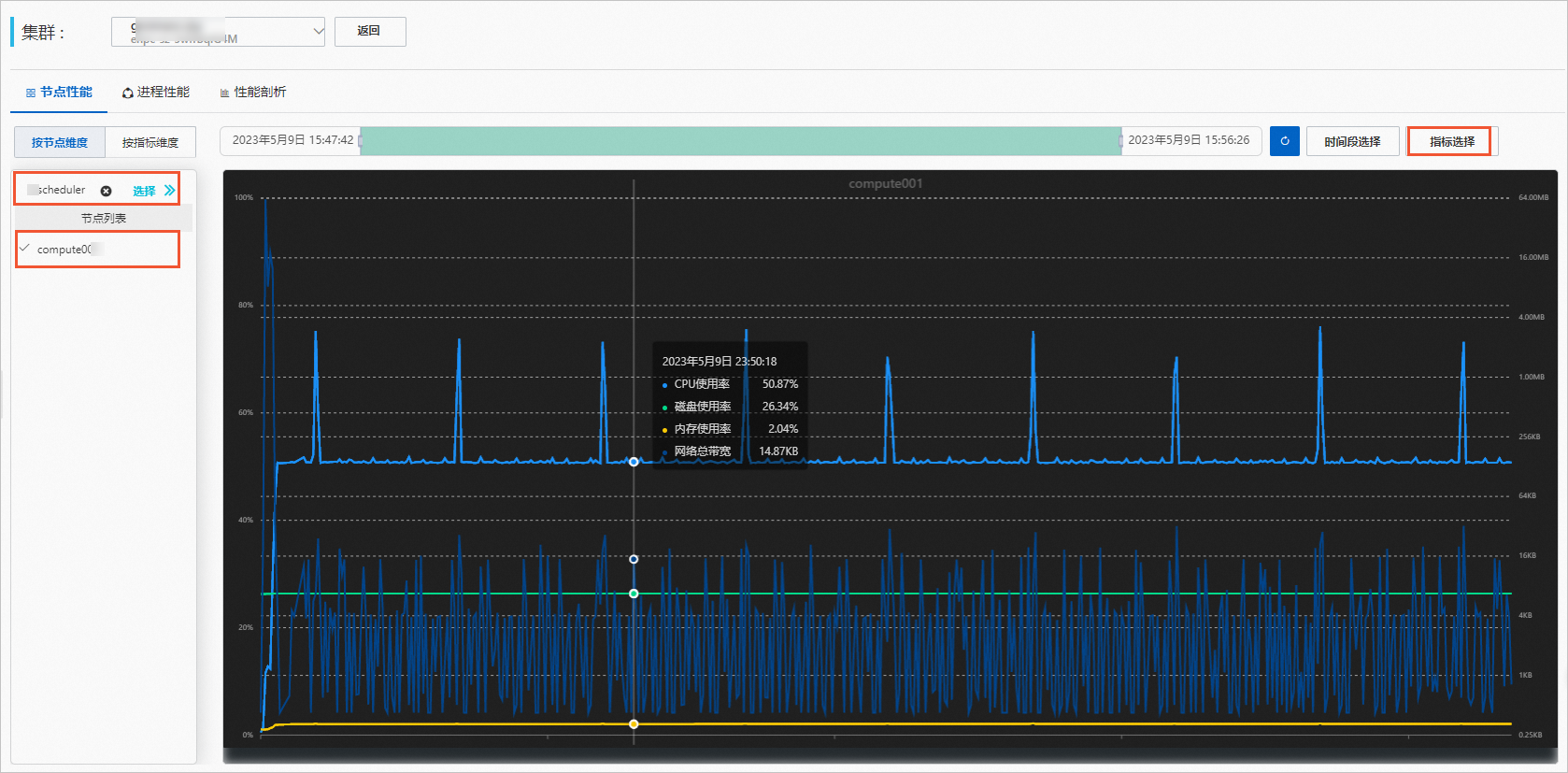
在节点性能页签下,查看节点性能。
选择作业和计算节点。
(可选)设置时间段。
选择作业后,时间段会自动调整为作业运行的时间段,您也可以自行调整时间段。
单击指标选择,选择要查看的指标。
查看节点性能图。
结果示例如下,鼠标上移到图形上方可以查看详细数据。
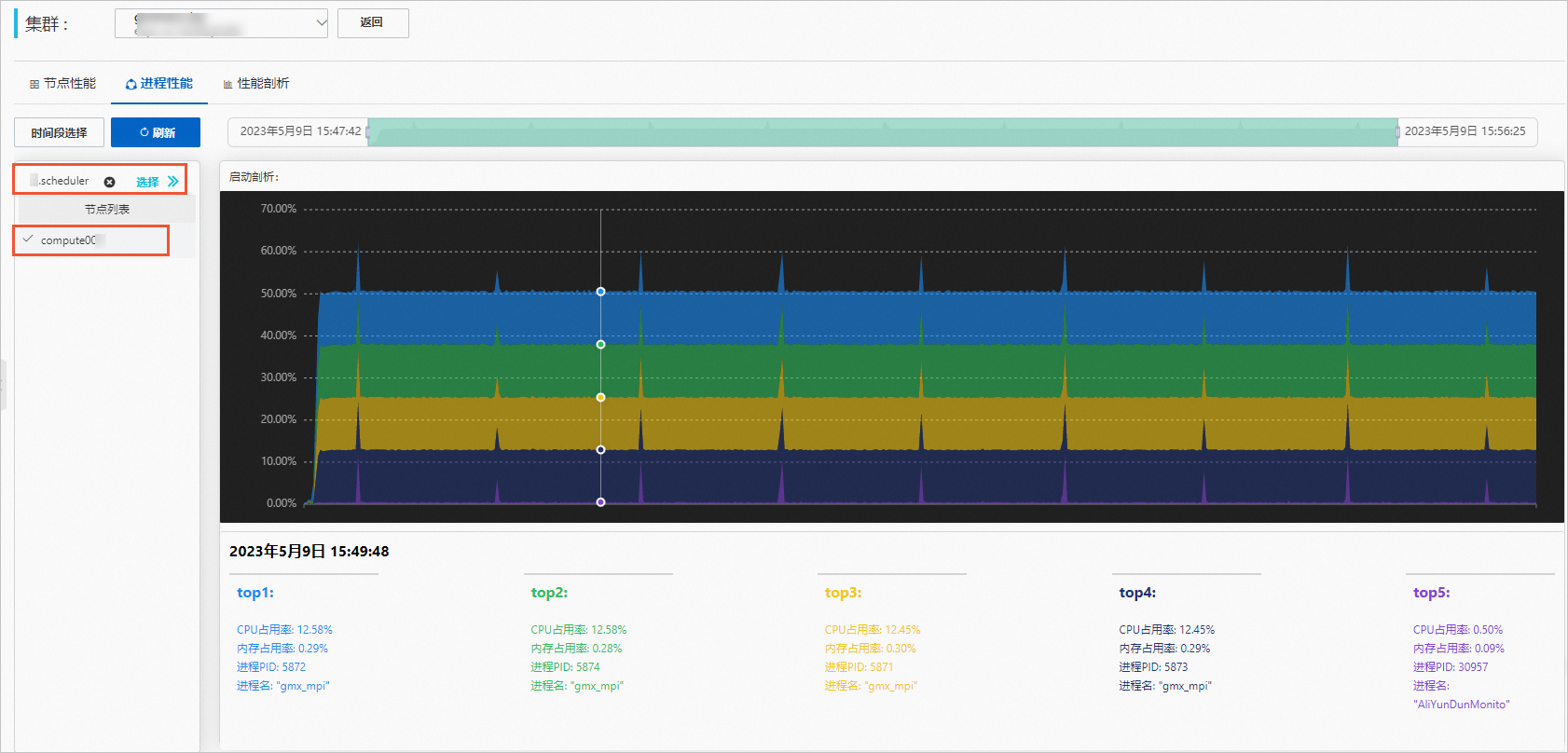
单击进程性能页签,查看进程性能。
选择作业和计算节点。
(可选)设置时间段。
选择作业后,时间段会自动调整为作业运行的时间段,您也可以自行调整时间段。
查看进程性能图。
结果示例如下,鼠标上移到图形上方可以查看详细数据。
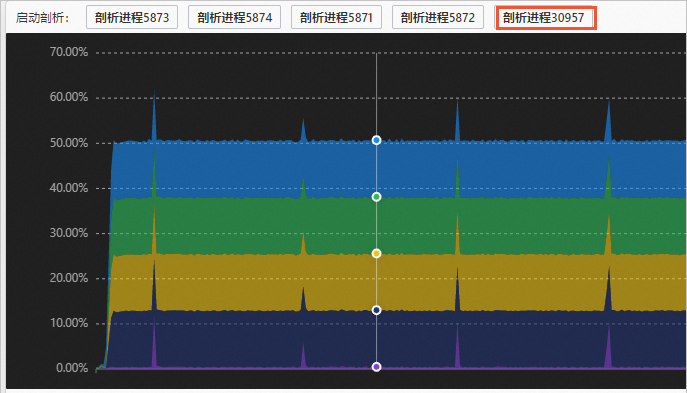
启动性能剖析任务。
在进程性能图上单击您想要的时间点,然后在图示上方单击您想要剖析的进程。
在弹出的对话框中设置性能剖析参数,然后单击确定。
参数包括时长和采样频率,单击确定后即可启动对作业的实时性能剖析。
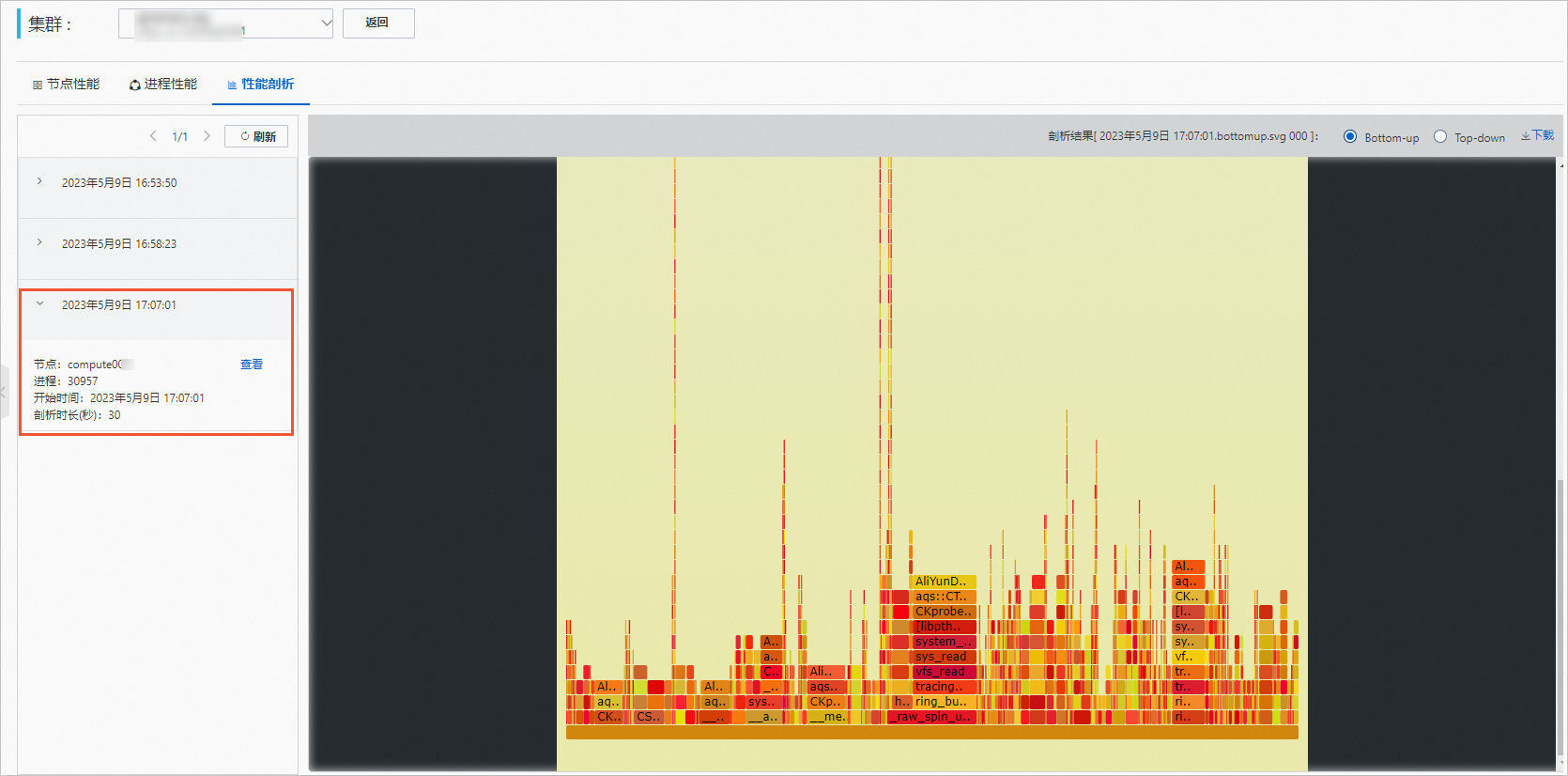
单击性能剖析页签,查看剖析结果。
选择剖析任务,单击查看。
查看剖析结果。
结果示例如下,鼠标上移到图形上方可以查看详细数据。