
本文以Amber软件为例介绍如何在E-HPC上进行分子动力学模拟。
背景信息
Amber(Assisted Model Building with Energy Refinement)是一款开源的广泛使用的分子动力学模拟软件,主要用于研究分子系统在生物化学领域的动态性质。它由多个程序组成,可以协调工作,广泛应用于蛋白质、核酸、糖等生物大分子的计算模拟。Amber软件的核心动力学引擎开发语言为Fortran90,支持多CPU并行运算与GPU加速,适合于进行大规模的分子动力学模拟。
准备工作
创建一台具有NVIDIA GPU卡的ECS实例,例如GPU计算型实例规格族gn7i等。更多信息,请参见自定义购买实例和GPU云服务器(gn/vgn/sgn系列)。
本文示例配置如下:
实例规格:ecs.gn7i-c8g1.2xlarge
操作系统:Alibaba Cloud Linux 3
建议您将实例部署在与集群相同的地域内,以免增加后续操作。
步骤一:安装并配置软件
远程连接已创建的ECS实例。具体操作,请参见通过密码或密钥认证登录Linux实例。
执行以下命令,安装OSS工具。
sudo yum install -y unzip sudo -v ; curl https://gosspublic.alicdn.com/ossutil/install.sh | sudo bash ossutil执行以下命令,修改
ossutilconfig配置文件,您可以设置OSS的Endpoint、AccessKeyID、AccessKeySecret等信息。更多信息,请参见配置ossutil。vi ~/.ossutilconfig执行以下命令,安装所需的开发工具和库文件。
sudo yum install -y gcc gcc-c++ make automake tcsh gcc-gfortran which flex bison patch bc libXt-devel libXext-devel perl perl-ExtUtils-MakeMaker util-linux wget bzip2 bzip2-devel zlib-devel tar执行以下命令,安装unrar工具。
sudo wget https://www.rarlab.com/rar/rarlinux-x64-612.tar.gz --no-check-certificate sudo tar -zxvf rarlinux-x64-612.tar.gz执行以下命令,安装CMake。
cd /usr/local/ sudo wget https://github.com/Kitware/CMake/releases/download/v3.18.1/cmake-3.18.1-Linux-x86_64.sh chmod +x cmake-3.18.1-Linux-x86_64.sh ./cmake-3.18.1-Linux-x86_64.sh export PATH=$PATH:/usr/local/cmake-3.18.1-Linux-x86_64/bin sudo yum install -y gcc gcc-c++ make automake升级GCC。
执行以下命令,安装CentOS Software Collections Release Repository的RH版本。
sudo yum install centos-release-scl-rh -y执行以下命令,修改
CentOS-SCLo-scl-rh.repo文件。sudo vi /etc/yum.repos.d/CentOS-SCLo-scl-rh.repo执行以下命令,安装Developer Toolset 7。
sudo yum install devtoolset-7 -y说明请根据您的项目需求和依赖环境,选取合适的Developer Toolset版本进行安装,以确保开发环境的兼容性和最优性能。
Devtoolset-3旨在配合GCC 4.x.x系列版本,为早期项目提供支持。
Devtoolset-4与GCC 5.x.x版本集成,适配中期软件栈的开发要求。
Devtoolset-6精确匹配GCC 6.x.x系列,引入了更多现代语言特性支持。
Devtoolset-7则是专为GCC 7.x.x版本设计,集成了最先进的编译技术和优化策略。
执行以下命令,设置环境变量。
source /opt/rh/devtoolset-7/enable
安装NVIDIA驱动和SDK。
重要鉴于NVIDIA图形处理器(GPU)与CUDA软件平台之间的紧密耦合性,为确保最佳性能并减少潜在的兼容性问题,建议将NVIDIA GPU驱动程序与CUDA工具包作为独立步骤进行部署。
执行以下命令,安装NVIDIA驱动。更多信息,请参见安装CUDA。
sudo wget https://us.download.nvidia.com/tesla/418.226.00/nvidia-driver-local-repo-rhel7-418.226.00-1.0-1.x86_64.rpm rpm -i nvidia-driver-local-repo-rhel7-418.226.00-1.0-1.x86_64.rpm sudo yum clean all sudo yum install cuda-drivers sudo reboot执行以下命令,安装NVIDIA的SDK。
sudo wget https://developer.download.nvidia.com/compute/cuda/10.1/Prod/local_installers/cuda_10.1.243_418.87.00_linux.run chmod +x cuda_10.1.243_418.87.00_linux.run ./cuda_10.1.243_418.87.00_linux.run --toolkit --samples --silent
编译Open MPI。
执行以下命令,下载版本为3.0.0的Open MPI源代码压缩包。
curl -O -L https://download.open-mpi.org/release/open-mpi/v3.0/openmpi-3.0.0.tar.gz依次执行以下命令,配置OpenMPI以支持CUDA。
export PATH=/usr/local/cuda/bin/:$PATH sudo tar zxvf openmpi-3.0.0.tar.gz cd openmpi-3.0.0/ ./configure --prefix=/opt/openmpi/ --with-cuda=/usr/local/cuda/执行以下命令进行编译。
sudo make -j 8 all sudo make -j 8 install执行以下命令,配置环境变量。
export PATH=/usr/local/cuda/bin/:/opt/openmpi/bin:$PATH
配置Amber软件。
进入Amber官网,下载
AmberTools24.tar.bz2与Amber24.tar.bz2文件,并在/opt/目录下进行解压缩操作。执行以下命令,修改Amber编译参数。
cd /opt/amber24_src/build/ DMPI=TRUE openmpiDCUDA=TRUE依次执行以下命令,运行编译并完成安装。
sudo ./run_cmake sudo make install依次执行以下命令,测试Amber。
source /opt/amber24/amber.sh make test.serial
打包编译好的OpenMPI和Amber软件。
创建目录。
cd mkdir amber-openmpi-package cd amber-openmpi-package使用
cp命令,将编译好的OpenMPI和Amber的可执行文件复制到amber-openmpi-package目录中。执行以下命令,创建压缩包。
cd tar -czf amber-openmpi-package.tar.gz amber-openmpi-package/
步骤二:创建自定义镜像
关于如何创建自定义镜像,请参见使用实例创建自定义镜像。
步骤三:使用自定义镜像创建E-HPC集群
创建一个E-HPC集群。具体操作,请参见创建标准版集群。
本文使用的集群配置示例如下:
配置项
配置
系列
标准版
部署模式
公共云集群
集群类型
SLURM
节点配置
包含1个管理节点和2个计算节点,规格如下:
管理节点:采用ecs.r7.xlarge实例规格,该规格配置为4 vCPU,32 GiB内存。
说明请根据实际业务需求,调整管理节点的实例规格,以提高或降低资源配置。
计算节点:采用ecs.gn6i-c24g1.12xlarge实例规格,该规格配置为48 vCPU、186 GiB内存。
集群镜像
镜像配置选择本文中已创建的自定义镜像,包括集群中的管理节点、登录节点和计算节点。
创建一个集群用户。具体操作,请参见用户管理。
集群用户用于登录集群,进行编译软件、提交作业等操作。本文创建的用户示例如下:
用户名:testuser
用户组:普通权限组
(可选)开启集群自动伸缩功能。具体操作,请参见自动伸缩节点。
说明您可以通过集群的弹性伸缩能力,无需手动操作,即可实现计算节点的动态分配,根据实时负载自动调整计算节点数量,以提升资源管理效率。
步骤四:创建MOE可视化任务
使用已创建的
testuser用户登录E-HPC Portal。具体操作,请参见登录E-HPC Portal。
单击右上角
 图标,使用Workbench远程连接集群登录节点。
图标,使用Workbench远程连接集群登录节点。将编译好的OpenMPI和Amber软件解压缩到
/opt目录中,确保所有节点可以共享使用。sudo tar -xzf amber-openmpi-package.tar.gz -C /opt安装MOE。
说明MOE是一款商业软件,您需要自行购买,然后遵循MOE官方指南进行安装。
设置环境变量。
执行以下命令,创建可执行文件
deploy_amber_openmpi.sh。vim deploy_amber_openmpi.sh将以下内容写入并保存退出。
#!/bin/bash # 设置 MOE 环境变量 export MOE=/opt/moe_2019.0102 export LD_LIBRARY_PATH=$MOE/lib:$LD_LIBRARY_PATH export PATH=$MOE/bin-lnx64:/usr/local/cuda/bin/:/opt/openmpi/bin:$PATH export AMBERHOME=/opt/amber24依次执行以下命令,顺利执行脚本文件。
chmod +x deploy_amber_openmpi.sh ./deploy_amber_openmpi.sh
创建MOE可视化任务。
在E-HPC Portal页面顶部导航栏,选择任务管理。
在页面上方,单击moe。
在创建作业页面,填写以下参数信息。
参数
说明
会话名称
运行moe访问集群产生的会话的名称。
运行节点
运行moe的节点。
如果选择队列,会自动分配队列中的节点。
如果选择节点,目前仅支持localhost节点,即为当前节点。
CPU核数
运行moe所使用的CPU核数。
分辨率
可视化界面的分辨率。
应用启动命令
保持默认为
moe。单击提交作业。
进入可视化界面。
在E-HPC Portal页面顶部导航栏,选择会话管理。
按照会话名称找到moe会话,单击会话名称。
进入E-HPC集群可视化界面进行操作。
步骤五:提交作业
本文以测试算例压缩包 test.rar 和作业执行脚本 run.sh文件为例来说明如何提交作业。在实际操作中,请根据您的具体业务需求,替换成相应的作业文件。
在E-HPC Portal页面,单击右上角
图标,使用Workbench远程连接集群登录节点。执行以下命令,解压缩测试用例
test.rar文件。unrar x test.rar执行以下命令,修改
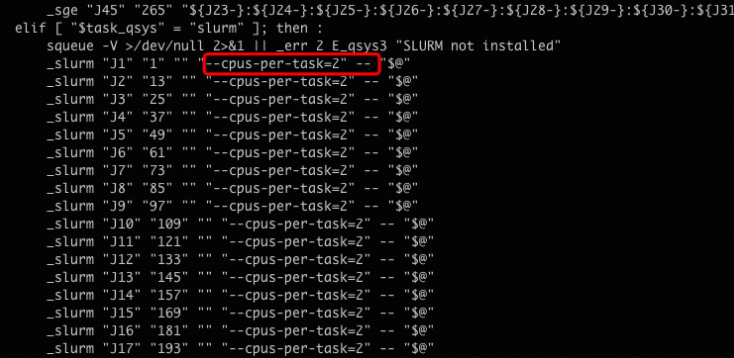
run.sh文件,限定每个作业需要2个CPU核心。cd test/ vim run.sh修改示例如下所示:
执行以下命令,提交作业。
./run.sh -qsys slurm -submit
步骤六:查看作业结果
执行以下任一命令,查询作业详情。
Slurm Sacct命令
sacctMOE命令
./run.sh -qsys slurm -status